四川大学贾大团队连发两文揭示囊泡运输在脑桥小脑发育不全症中的致病机制
作者:刘哲(四川大学) 发布时间:2020/6/1 8:00:00
脑桥小脑发育不全(Pontocerebellar hypoplasia,PCH)是一组涉及脑发育的神经系统疾病,该病会对患者的脑桥和小脑功能造成严重损害。患者通常幼年发病且病情随病程的发展日渐加重,病征主要表现为头小畸形、整体发育延迟、行动不便以及轻度至重度智力障碍[1,2]。迄今为止,已经发现了十几个基因与PCH的发生和发展有关。最近,通过对病人基因组测序在TBC1D23上发现了PCH的新型致病突变[3-5]。在哺乳动物细胞中,TBC1D23定位于高尔基体上。通过与内吞体上蛋白质FAM21结合,TBC1D23将内吞体囊泡募集到高尔基体上,从而介导从内吞体到高尔基体的囊泡运输[6,7]。但是,TBC1D23突变导致PCH的分子机制还并不清楚。
2019年10月,四川大学贾大课题组在PNAS上发表了题为Structural and functional studies of TBC1D23 C-terminal domain provide a link between endosomal trafficking and PCH的长文,证实囊泡运输的缺馅是导致PCH的重要原因,在探索PCH的分子机制上迈出了重要的一步。近日,该团队在PLoS Biology上发表题为Structure of TBC1D23 N-terminus reveals a novel role forrhodanese domain的文章, 进一步完善了TBC1D23突变在PCH发生中的作用机制 。

在PNAS一文中,作者发现tbc1d23敲减的斑马鱼能较好地模拟携带TBC1D23突变体病人在神经发育和运动机能上的多种表型。TBC1D23基因突变导致在这些病人身上只表达C端结构域缺失的蛋白。利用斑马鱼模型,作者发现Tbc1d23的C端结构域在脑和神经发育起着不可或缺的作用(图1)。作者还发现Tbc1d23在细胞中囊泡运输的功能与其在斑马鱼神经发育中的功能正相关,因此提出囊泡运输的缺陷是 TBC1D23 突变引起PCH的内在机理 (图2) 。基于该研究的新颖性,囊泡领域知名专家Dr. Peter Cullen (英国University of Bristol教授、Welcome Trust Investigator)专门在Twitter发文,称赞本研究是“ Really informative study from Dr. Da Jia’s lab ” (图3)。
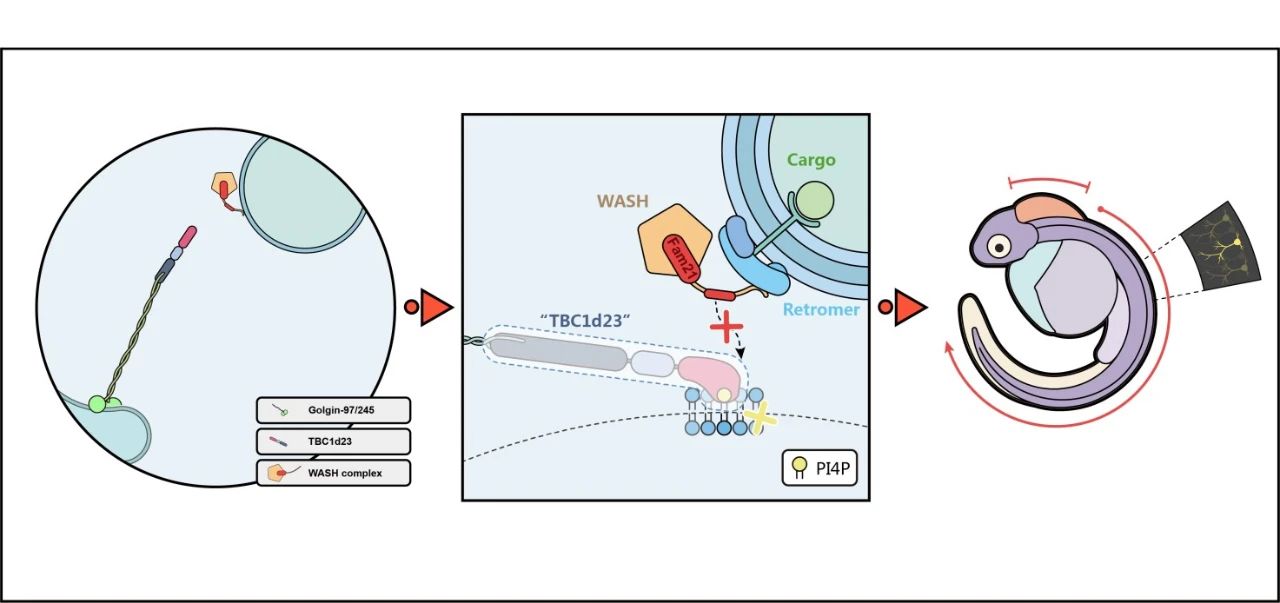
图2 TBC1D23突变在PCH发生中的作用机制
图3 Peter Cullen教授对PNAS上发表结果的点评
在PLoS Biology一文中,作者进一步研究了TBC1D23的N端的结构与功能。TBC1D23的N端由TBC和rhodanese结构域组成。其TBC和经典的TBC结构域不同,没有RabGAP的活性;根据蛋白质序列预测,TBC1D23的rhodanese可能具有蛋白质磷酸酶或者硫转移酶活性[6]。作者则发现TBC1D23的rhodanese结构域并不具有酶活性。该rhodanese与TBC结构域一起发挥作用,与高尔基蛋白Golgin-97/245互作。该互作对于TBC1D23的高尔基体定位十分关键。与体外实验一致,作者在斑马鱼上发现在Tbc1d23与Golgin-97/245互作的位点对于斑马鱼的轴突和HuC神经元发育是必须的(图4),但rhodanese结构域的“酶活位点”却可有可无。

图4 Tbc1d23与Golgin-97/245互作对于斑马鱼的轴突和HuC神经元发育是必须
综上,对于TBC1D23的这两项深入研究,为进一步探讨PCH的发生机制及开发治疗手段奠定了基础。
四川大学的黄文杰、刘哲、阳繁及上海高等研究院的周欢为PNAS论文的共同第一作者,贾大为该论文的通讯作者。四川大学的刘定东、阳繁、刘哲、王瑾瑞为PLoS Biology论文的共同第一作者,贾大、莫显明、孙庆祥为该论文的通讯作者。四川大学的魏于全、张小虎,西南大学的李礼,华中科技大学的马聪等为课题提供了大力支持。
原文链接:
https://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.3000746
https://www.pnas.org/content/early/2019/10/14/1909316116
参考文献:
1. Rudnik-Schoneborn, S., Barth, P.G. & Zerres, K. Pontocerebellar hypoplasia. Am J Med Genet C Semin Med Genet 166C, 173-83 (2014).2. Namavar, Y., Barth, P.G., Poll-The, B.T. & Baas, F. Classification, diagnosis and potential mechanisms in pontocerebellar hypoplasia. Orphanet J Rare Dis 6, 50 (2011).
3. Marin-Valencia, I. et al. Homozygous Mutations in TBC1D23 Lead to a Non-degenerative Form of Pontocerebellar Hypoplasia. Am J Hum Genet 101, 441-450 (2017).
4. Ivanova, E.L. et al. Homozygous Truncating Variants in TBC1D23 Cause Pontocerebellar Hypoplasia and Alter Cortical Development. Am J Hum Genet 101, 428-440 (2017).
5. Harripaul, R. et al. Mapping autosomal recessive intellectual disability: combined microarray and exome sequencing identifies 26 novel candidate genes in 192 consanguineous families. Mol Psychiatry 23, 973-984 (2018).
6. Shin, J.J.H., Gillingham, A.K., Begum, F., Chadwick, J. & Munro, S. TBC1D23 is a bridging factor for endosomal vesicle capture by golgins at the trans-Golgi. Nat Cell Biol 19, 1424-1432 (2017).
7. Navarro Negredo, P., Edgar, J.R., Manna, P.T., Antrobus, R. & Robinson, M.S. The WDR11 complex facilitates the tethering of AP-1-derived vesicles. Nat Commun 9, 596 (2018).