李原/王强/蔡涛组合作鉴定抗氧化基因OXR1突变感音性耳聋遗传家系
作者:CZRC 发布时间:2022/9/28 9:00:00
耳聋是人类最常见的感觉障碍之一,全世界约有3.6亿人受到影响。近千分之一的新生儿患有先天性听力损伤,50%-60%的新生儿听力障碍由遗传因素所致。目前已经克隆的与遗传性耳聋相关的基因已超过150个[1-3]。在临床表型上大约30-40%的遗传性耳聋是综合征型,60-70%为非综合征型耳聋[4]。2006年第二次全国残疾人抽样调查显示,我国有2780万人存在听力和语言障碍[5],每年有近30000名新生儿患有先天性感音神经性耳聋(sensorineural hearing loss,SNHL)[6,7]。然而,先天性耳聋的病因复杂而且多样,仍有相当一部分遗传性耳聋病理性突变是由罕见的基因突变所致。这些罕见突变仅在单个或几个家族中检测发现[8]。因此,发现并且鉴定新的耳聋基因变异,将有助于丰富遗传性感音性耳聋的基因谱,为聋哑孕检提供理论依据及遗传性耳聋基因治疗提供理论基础。
2022年09月22日,中日友好医院耳鼻喉头颈外科李原团队、华南理工大学医学院王强团队、美国国立卫生研究院(NIH)牙科和颅面研究所(NIDCR)蔡涛团队,协同合作在国际经典遗传学期刊《Human Molecular Genetics》在线发表了题为“A novel recessive mutation in OXR1 is identified in patient with hearing loss recapitulated by the knockdown zebrafish”的研究论文。该研究对354个先天性耳聋家庭中389名成员进行了全外显子组测序(Whole Exome Sequencing,WES)分析。首次在一名4岁的感音神经性耳聋女孩中鉴定出了抗氧化基因1 (oxidation resistance gene 1, OXR1)纯合子错义突变(c.233A>G, p.Lys78Arg)。先证者是非近亲结婚的健康中国父母的第二个孩子(图1A )。她是剖腹产出生的早产儿,出生时体重3000克,身长50厘米。由于未能通过新生儿听力筛查,分别于6个月龄、1岁和2岁时经主、客观听力学检查诊断为双侧重度感音神经性耳聋(SNHL)(图1B)。对其家庭成员进行WES家系分析发现,先天性耳聋女孩为OXR1基因纯合错义突变(图2A)。其父母和姐姐为OXR1基因的杂合突变携带者。该变异极其罕见( p= 0.000184)。对患者家乡采集的50例当地居民进行正常对照的Sanger测序,均为野生型,未发现OXR1基因类似突变。
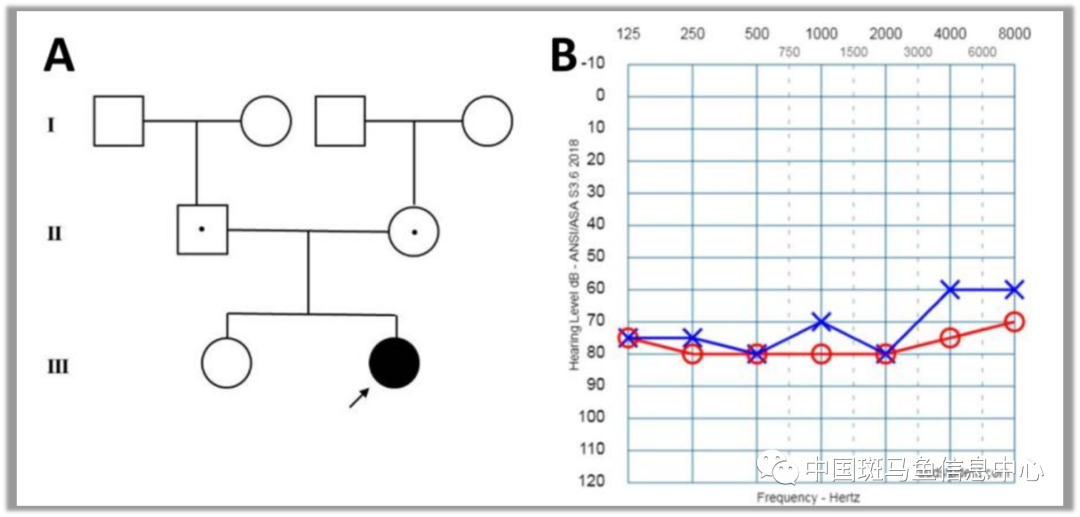
图1 (A)先证者3代家系图。(B)纯音测听显示双侧重度感音神经性耳聋,在最大声刺激下听性脑干反应(ABR )缺失。
图1 (A)先证者3代家系图。(B)纯音测听显示双侧重度感音神经性耳聋,在最大声刺激下听性脑干反应(ABR )缺失。
OXR1基因从斑马鱼到人类在进化上高度保守。为了研究OXR1在听觉器官发育中的作用,验证OXR1基因突变的致聋性,研究者利用斑马鱼模型对其进行了探究。本研究发现斑马鱼同源基因oxr1b(人类OXR1的一个同源基因)在斑马鱼的听觉平衡神经节(statoacoustic ganglions, SAG)和斑马鱼后侧线神经节(posterior lateral line ganglions, pLL)中高表达(图2A, 2B)。斑马鱼中oxr1b的敲降导致了SAG和pLL的显著发育缺陷(图2C)。
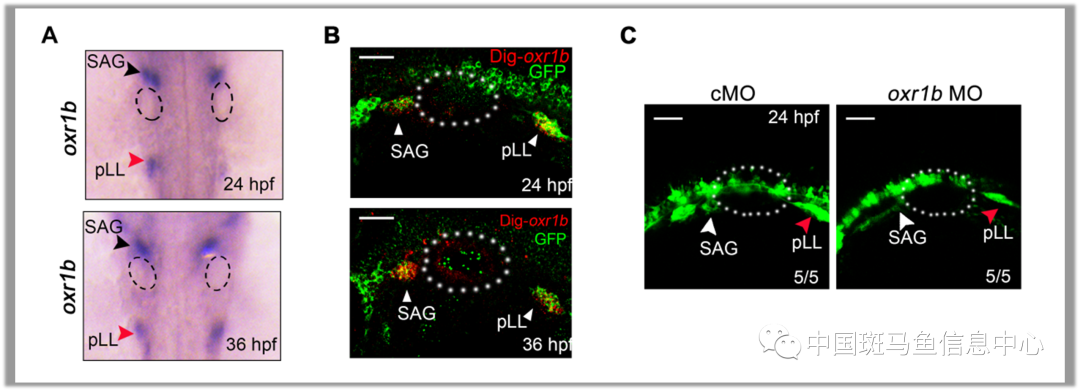
图2 (A-B) oxr1b 在斑马鱼24 hpf和36 hpf胚胎的听觉平衡神经节 (SAG) 和后外侧线神经节 (pLL) 有特异性表达。(C)oxr1b是SAG发育所必需的。
图2 (A-B) oxr1b 在斑马鱼24 hpf和36 hpf胚胎的听觉平衡神经节 (SAG) 和后外侧线神经节 (pLL) 有特异性表达。(C)oxr1b是SAG发育所必需的。
为了验证因c.233A>G突变导致的OXR1相关听力损失,通过联合注射野生型OXR1 mRNA进行救援实验,mRNA在很大程度上挽救了MO诱导的表型,而c.233A>G突变型OXR1 mRNA不能修复SAG缺陷(图3)。作为一个新的候选基因,OXR1的额外突变应该从较大的听力损失人群中进行筛查。
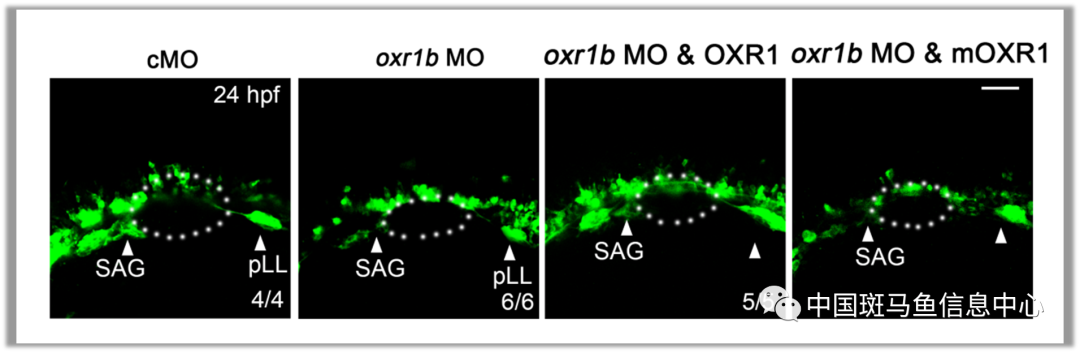
图3 oxr1b失活诱导的SAG缺陷可以通过过表达野生型人OXR1而非突变型OXR1 ( mOXR1 , c.233A > G)来挽救。
图3 oxr1b失活诱导的SAG缺陷可以通过过表达野生型人OXR1而非突变型OXR1 ( mOXR1 , c.233A > G)来挽救。
OXR1相关通路分析发现TBC1D24是OXR1的一个含有TLDc结构域的同源基因,以前在听力损失患者中被发现。有趣的是,在患者或动物模型中,OXR1相互作用分子如ATP6V1B1和ESR1的突变或敲除也与听力损失相关,提示OXR1及其相关蛋白在耳蜗发育和听觉功能中的重要作用。后续的研究则可以通过CRISPR/Cas9基因组编辑技术,建立一个稳定的突变体,以此来探索潜在的分子OXR1在听觉相关通路中的作用机制。
中日友好医院耳鼻喉科头颈外科主任医师李原为论文第一作者,中国科学院动物研究所助理研究员宁国柱博士(现单位为广东医科大学附属医院)为论文共同第一作者。美国国立卫生研究院牙科和颅面研究所研究员蔡涛,华南理工大学医学院王强教授为共同通讯作者。该研究作为一项公益性研究,得到了中国听力学发展基金会(NO5310000500017757XA19013)和北京安琪尔基因医学科技有限公司的支持。本研究还得到了国家自然科学基金(32025014和31872838)和国家重点研发专项(2018YFA0800200和2020YFA0804000)的支持。此研究感谢参与这项研究的耳聋病人及其家人,感谢南通大学刘东教授和巩杰副教授在实验技术上的帮助。
参考文献
1. Cryns, K. and Van Camp, G. (2004) Deafness genes and their diagnostic applications. Audiol Neurootol, 9, 2-22.
2. Petit, C., Levilliers, J. and Hardelin, J.P. (2001) Molecular genetics of hearing loss. Annu Rev Genet, 35, 589-646.
3. Raviv, D., Dror, A.A. and Avraham, K.B. (2010) Hearing loss: a common disorder caused by many rare alleles. Ann N Y Acad Sci, 1214, 168-179.
4. Tekin, M., Arnos, K.S. and Pandya, A. (2001) Advances in hereditary deafness. Lancet, 358, 1082-1090.
5. He, Z.H., Li, M., Zou, S.Y., Liao, F.L., Ding, Y.Y., Su, H.G., Wei, X.F., Wei, C.J., Mu, Y.R. and Kong, W.J. (2019) Protection and Prevention of Age-Related Hearing Loss. Adv Exp Med Biol, 1130, 59-71.
6. Dai P, L.X., Yu F, Zhu Q. (2006) Molecular etiology of patients with nonsyndromic hearing loss from deaf-mute schools in 18 provinces of China. Chin J Otol 4: 1–5.
7. Sun, X.B., Wei, Z.Y., Yu, L.M., Wang, Q. and Liang, W. (2008) [Prevalence and etiology of people with hearing impairment in China]. Zhonghua Liu Xing Bing Xue Za Zhi, 29, 643-646.
8. Diaz-Horta, O., Duman, D., Foster, J., 2nd, Sirmaci, A., Gonzalez, M., Mahdieh, N., Fotouhi, N., Bonyadi, M., Cengiz, F.B., Menendez, I. et al. (2012) Whole-exome sequencing efficiently detects rare mutations in autosomal recessive nonsyndromic hearing loss. PLoS One, 7, e50628.